Demo: Spatial data from Slide-seq#
This demo uses data from:
Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution
Rodriques et al (Science 2019)
For this demo, you need the data files for Puck_180819_12
Taken from: https://portals.broadinstitute.org/single_cell/study/SCP354/slide-seq-study#study-summary
Specifically these two files:
MappedDGEForR.csv
BeadMapping_10-17_1014/BeadLocationsForR.csv
For convenience, we have wrapped this in an AnnData object that you will download below.
Covered Here#
Computing Autocorrelation in Hotspot to identify spatial genes
Computing local correlations between spatial genes to identify modules
Plotting modules, correlations, and module scores
Not Covered Here#
Data preprocessing (see Squidpy/Scanpy)
import sys
#if branch is stable, will install via pypi, else will install from source
branch = "anndata"
IN_COLAB = "google.colab" in sys.modules
if IN_COLAB:
!pip install --quiet --upgrade numba
!pip install git+https://github.com/yoseflab/hotspot@$branch#egg=hotspot
!pip install --quiet scanpy
!pip install --quiet mplscience
|████████████████████████████████| 3.3 MB 8.3 MB/s
|████████████████████████████████| 34.5 MB 1.4 MB/s
?25hCollecting hotspot
Cloning https://github.com/yoseflab/hotspot (to revision anndata) to /tmp/pip-install-6tu2qe3n/hotspot_3d0fc0f8c2b94342ad19e622464099fe
Running command git clone -q https://github.com/yoseflab/hotspot /tmp/pip-install-6tu2qe3n/hotspot_3d0fc0f8c2b94342ad19e622464099fe
Running command git checkout -b anndata --track origin/anndata
Switched to a new branch 'anndata'
Branch 'anndata' set up to track remote branch 'anndata' from 'origin'.
Requirement already satisfied: matplotlib>=3.0.0 in /usr/local/lib/python3.7/dist-packages (from hotspot) (3.2.2)
Requirement already satisfied: numba>=0.43.1 in /usr/local/lib/python3.7/dist-packages (from hotspot) (0.55.1)
Requirement already satisfied: numpy>=1.16.4 in /usr/local/lib/python3.7/dist-packages (from hotspot) (1.21.5)
Requirement already satisfied: seaborn>=0.9.0 in /usr/local/lib/python3.7/dist-packages (from hotspot) (0.11.2)
Requirement already satisfied: scipy>=1.2.1 in /usr/local/lib/python3.7/dist-packages (from hotspot) (1.4.1)
Requirement already satisfied: pandas>=0.24.0 in /usr/local/lib/python3.7/dist-packages (from hotspot) (1.3.5)
Requirement already satisfied: tqdm>=4.32.2 in /usr/local/lib/python3.7/dist-packages (from hotspot) (4.62.3)
Requirement already satisfied: statsmodels>=0.9.0 in /usr/local/lib/python3.7/dist-packages (from hotspot) (0.10.2)
Requirement already satisfied: scikit-learn>=0.21.2 in /usr/local/lib/python3.7/dist-packages (from hotspot) (1.0.2)
Collecting anndata>=0.7
Downloading anndata-0.7.8-py3-none-any.whl (91 kB)
|████████████████████████████████| 91 kB 4.3 MB/s
?25hRequirement already satisfied: importlib_metadata>=0.7 in /usr/local/lib/python3.7/dist-packages (from anndata>=0.7->hotspot) (4.11.0)
Requirement already satisfied: xlrd<2.0 in /usr/local/lib/python3.7/dist-packages (from anndata>=0.7->hotspot) (1.1.0)
Requirement already satisfied: h5py in /usr/local/lib/python3.7/dist-packages (from anndata>=0.7->hotspot) (3.1.0)
Requirement already satisfied: packaging>=20 in /usr/local/lib/python3.7/dist-packages (from anndata>=0.7->hotspot) (21.3)
Requirement already satisfied: natsort in /usr/local/lib/python3.7/dist-packages (from anndata>=0.7->hotspot) (5.5.0)
Requirement already satisfied: zipp>=0.5 in /usr/local/lib/python3.7/dist-packages (from importlib_metadata>=0.7->anndata>=0.7->hotspot) (3.7.0)
Requirement already satisfied: typing-extensions>=3.6.4 in /usr/local/lib/python3.7/dist-packages (from importlib_metadata>=0.7->anndata>=0.7->hotspot) (3.10.0.2)
Requirement already satisfied: pyparsing!=2.0.4,!=2.1.2,!=2.1.6,>=2.0.1 in /usr/local/lib/python3.7/dist-packages (from matplotlib>=3.0.0->hotspot) (3.0.7)
Requirement already satisfied: cycler>=0.10 in /usr/local/lib/python3.7/dist-packages (from matplotlib>=3.0.0->hotspot) (0.11.0)
Requirement already satisfied: kiwisolver>=1.0.1 in /usr/local/lib/python3.7/dist-packages (from matplotlib>=3.0.0->hotspot) (1.3.2)
Requirement already satisfied: python-dateutil>=2.1 in /usr/local/lib/python3.7/dist-packages (from matplotlib>=3.0.0->hotspot) (2.8.2)
Requirement already satisfied: setuptools in /usr/local/lib/python3.7/dist-packages (from numba>=0.43.1->hotspot) (57.4.0)
Requirement already satisfied: llvmlite<0.39,>=0.38.0rc1 in /usr/local/lib/python3.7/dist-packages (from numba>=0.43.1->hotspot) (0.38.0)
Requirement already satisfied: pytz>=2017.3 in /usr/local/lib/python3.7/dist-packages (from pandas>=0.24.0->hotspot) (2018.9)
Requirement already satisfied: six>=1.5 in /usr/local/lib/python3.7/dist-packages (from python-dateutil>=2.1->matplotlib>=3.0.0->hotspot) (1.15.0)
Requirement already satisfied: threadpoolctl>=2.0.0 in /usr/local/lib/python3.7/dist-packages (from scikit-learn>=0.21.2->hotspot) (3.1.0)
Requirement already satisfied: joblib>=0.11 in /usr/local/lib/python3.7/dist-packages (from scikit-learn>=0.21.2->hotspot) (1.1.0)
Requirement already satisfied: patsy>=0.4.0 in /usr/local/lib/python3.7/dist-packages (from statsmodels>=0.9.0->hotspot) (0.5.2)
Requirement already satisfied: cached-property in /usr/local/lib/python3.7/dist-packages (from h5py->anndata>=0.7->hotspot) (1.5.2)
Building wheels for collected packages: hotspot
Building wheel for hotspot (setup.py) ... ?25l?25hdone
Created wheel for hotspot: filename=hotspot-0.9.1-py3-none-any.whl size=25699 sha256=32673675d4f8d6d438cf928d204c37d679c313816ab5132f7cd71ee8958ad92f
Stored in directory: /tmp/pip-ephem-wheel-cache-5a1kxpla/wheels/5f/06/1e/4c02025a303d8ff93d24e7c0d16a570a6b7da858ddf9680c63
Successfully built hotspot
Installing collected packages: anndata, hotspot
Successfully installed anndata-0.7.8 hotspot-0.9.1
|████████████████████████████████| 2.0 MB 8.5 MB/s
|████████████████████████████████| 86 kB 5.3 MB/s
|████████████████████████████████| 1.1 MB 64.6 MB/s
|████████████████████████████████| 63 kB 1.8 MB/s
?25h Building wheel for umap-learn (setup.py) ... ?25l?25hdone
Building wheel for pynndescent (setup.py) ... ?25l?25hdone
Building wheel for sinfo (setup.py) ... ?25l?25hdone
ERROR: pip's dependency resolver does not currently take into account all the packages that are installed. This behaviour is the source of the following dependency conflicts.
markdown 3.3.6 requires importlib-metadata>=4.4; python_version < "3.10", but you have importlib-metadata 1.7.0 which is incompatible.
import scanpy as sc
import hotspot
import numpy as np
import mplscience
import matplotlib
/usr/local/lib/python3.7/dist-packages/statsmodels/tools/_testing.py:19: FutureWarning: pandas.util.testing is deprecated. Use the functions in the public API at pandas.testing instead.
import pandas.util.testing as tm
url = "https://github.com/YosefLab/scVI-data/blob/master/rodriques_slideseq.h5ad?raw=true"
adata = sc.read("rodriques_slideseq.h5ad", backup_url=url)
adata.obs["total_counts"] = np.asarray(adata.X.sum(1)).ravel()
adata.layers["csc_counts"] = adata.X.tocsc()
# renormalize the data for expression viz on plots
sc.pp.normalize_total(adata)
sc.pp.log1p(adata)
Creating the Hotspot object#
To start an analysis, first creat the hotspot object When creating the object, you need to specify:
The gene counts matrix
Which background model to use
The latent space we are using to compute our cell metric
Here we use the physical barcode positions
The per-cell scaling factor
Here we use the number of umi per barcode
Once the object is created, the neighborhood graph must be computed with create_knn_graph
The two options that are specificied are n_neighbors which determines the size of the neighborhood, and weighted_graph.
Here we set weighted_graph=False to just use binary, 0-1 weights and n_neighbors=300 to create a local neighborhood size of the nearest 300 barcodes. Larger neighborhood sizes can result in more robust detection of correlations and autocorrelations at a cost of missing more fine-grained, smaller-scale, spatial patterns
# Create the Hotspot object and the neighborhood graph
hs = hotspot.Hotspot(
adata,
layer_key="csc_counts",
model='bernoulli',
latent_obsm_key="spatial",
umi_counts_obs_key="total_counts",
)
hs.create_knn_graph(
weighted_graph=False, n_neighbors=300,
)
Determining genes with spatial variation#
Now we compute autocorrelations for each gene, using the spatial metric, to determine which genes have the most spatial variation.
hs_results = hs.compute_autocorrelations(jobs=4)
hs_results.head()
100%|██████████| 6942/6942 [06:36<00:00, 17.53it/s]
| C | Z | Pval | FDR | |
|---|---|---|---|---|
| Gene | ||||
| Ttr | 0.198161 | 438.058454 | 0.0 | 0.0 |
| Plp1 | 0.110854 | 246.179606 | 0.0 | 0.0 |
| Enpp2 | 0.103074 | 211.241807 | 0.0 | 0.0 |
| Fth1 | 0.092905 | 207.076233 | 0.0 | 0.0 |
| Cartpt | 0.087053 | 180.180043 | 0.0 | 0.0 |
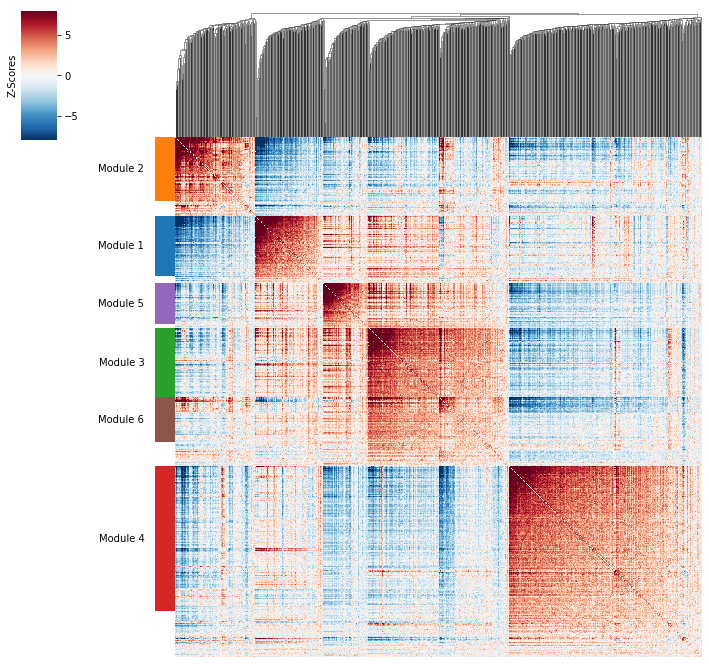
Grouping genes into spatial modules#
To get a better idea of what spatial patterns exist, it is helpful to group the genes into modules.
Hotspot does this using the concept of “local correlations” - that is,
correlations that are computed between genes between cells in the same neighborhood.
Here we avoid running the calculation for all Genes x Genes pairs and instead
only run this on genes that have significant spatial autocorrelation to begin with.
The method compute_local_correlations returns a Genes x Genes matrix of
Z-scores for the significance of the correlation between genes. This object
is also retained in the hs object and is used in the subsequent steps.
# Select the genes with significant spatial autocorrelation
hs_genes = hs_results.index[hs_results.FDR < 0.05]
# Compute pair-wise local correlations between these genes
lcz = hs.compute_local_correlations(hs_genes, jobs=4)
Computing pair-wise local correlation on 876 features...
100%|██████████| 876/876 [00:07<00:00, 110.16it/s]
100%|██████████| 383250/383250 [4:00:06<00:00, 26.60it/s]
Now that pair-wise local correlations are calculated, we can group genes into modules.
To do this, a convenience method is included create_modules which performs
agglomerative clustering with two caveats:
If the FDR-adjusted p-value of the correlation between two branches exceeds
fdr_threshold,
then the branches are not merged.If two branches are two be merged and they are both have at least
min_gene_thresholdgenes,
then the branches are not merged. Further genes that would join to the resulting merged module
(and are therefore ambiguous) either remain unassigned (ifcore_only=True) or are assigned to the module with the
smaller average correlations between genes, i.e. the least-dense module (ifcore_only=False)
The output is a Series that maps gene to module number. Unassigned genes are indicated with a module number of -1
This method was used to preserved substructure (nested modules) while still giving the analyst
some control. However, since there are a lot of ways to do hierarchical clustering, you can also
manually cluster using the gene-distances in hs.local_correlation_z
modules = hs.create_modules(
min_gene_threshold=20, core_only=False, fdr_threshold=0.05
)
modules.value_counts()
4 243
-1 163
3 117
2 108
1 101
6 75
5 69
Name: Module, dtype: int64
Plotting module correlations#
A convenience method is supplied to plot the results of hs.create_modules
hs.plot_local_correlations()
To explore individual genes, we can look at the genes with the top autocorrelation
in a given module as these are likely the most informative.
# Show the top genes for a module
module = 1
results = hs.results.join(hs.modules)
results = results.loc[results.Module == module]
results.sort_values('Z', ascending=False).head(10)
| C | Z | Pval | FDR | Module | |
|---|---|---|---|---|---|
| Gene | |||||
| Plp1 | 0.110854 | 246.179606 | 0.000000e+00 | 0.000000e+00 | 1.0 |
| Fth1 | 0.092905 | 207.076233 | 0.000000e+00 | 0.000000e+00 | 1.0 |
| Mbp | 0.059088 | 130.720208 | 0.000000e+00 | 0.000000e+00 | 1.0 |
| Apod | 0.026268 | 57.079755 | 0.000000e+00 | 0.000000e+00 | 1.0 |
| Mobp | 0.022971 | 50.674919 | 0.000000e+00 | 0.000000e+00 | 1.0 |
| Mal | 0.019166 | 41.898995 | 0.000000e+00 | 0.000000e+00 | 1.0 |
| Trf | 0.014862 | 32.176305 | 1.893429e-227 | 3.552483e-225 | 1.0 |
| Car2 | 0.012635 | 27.513785 | 6.004986e-167 | 9.925385e-165 | 1.0 |
| Cryab | 0.011001 | 23.612011 | 1.450564e-123 | 1.974473e-121 | 1.0 |
| Ermn | 0.010955 | 22.928582 | 1.205287e-116 | 1.494125e-114 | 1.0 |
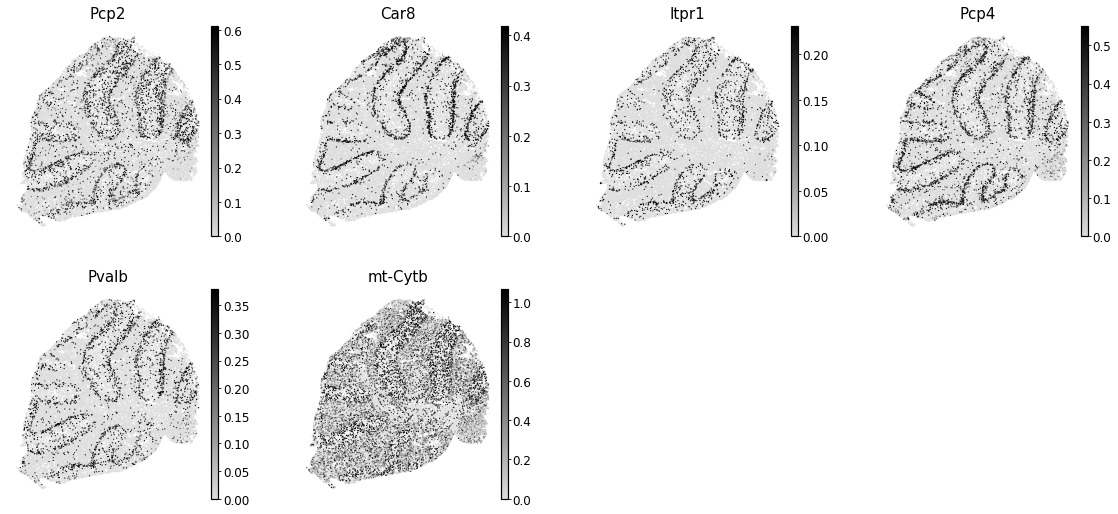
To get an idea of what spatial pattern the module is referencing, we can plot the top
module genes’ expression onto the spatial positions
cmap = matplotlib.colors.LinearSegmentedColormap.from_list(
'grays', ['#DDDDDD', '#000000'])
module = 2
results = hs.results.join(hs.modules)
results = results.loc[results.Module == module]
genes = results.sort_values('Z', ascending=False).head(6).index
with mplscience.style_context():
sc.pl.spatial(
adata,
color=genes,
cmap=cmap,
frameon=False,
vmin='p0',
vmax='p95',
spot_size=30,
)
Summary Module Scores#
To aid in the recognition of the general behavior of a module, Hotspot can compute
aggregate module scores.
module_scores = hs.calculate_module_scores()
module_scores.head()
Computing scores for 6 modules...
100%|██████████| 6/6 [01:24<00:00, 14.08s/it]
| 1 | 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|
| barcodes | ||||||
| CGTACAATTTTTTT | -0.421503 | 0.829196 | -0.025306 | -0.403850 | -0.209946 | 0.058708 |
| TTCGTTATTTTTTT | -0.433274 | 1.194746 | -0.251114 | -0.451753 | -0.206384 | 0.263886 |
| CAAACCAACCCCCC | -0.302593 | 0.609414 | -0.171035 | 0.265985 | -0.315975 | 0.350375 |
| TCTTTTCACCCCCC | -0.510236 | 1.309481 | -0.082019 | -0.743183 | -0.100390 | 0.198135 |
| GGATTGAACCCCCC | -0.554569 | 0.108010 | -0.132590 | -0.338540 | -0.142225 | 0.255825 |
module_cols = []
for c in module_scores.columns:
key = f"Module {c}"
adata.obs[key] = module_scores[c]
module_cols.append(key)
Here we can visualize these module scores by plotting them over the barcode positions:
with mplscience.style_context():
sc.pl.spatial(adata, color=module_cols, frameon=False, vmin="p0", vmax="p99", spot_size=30)
